Voici une courte présentation des conférences et ateliers du dimanche.
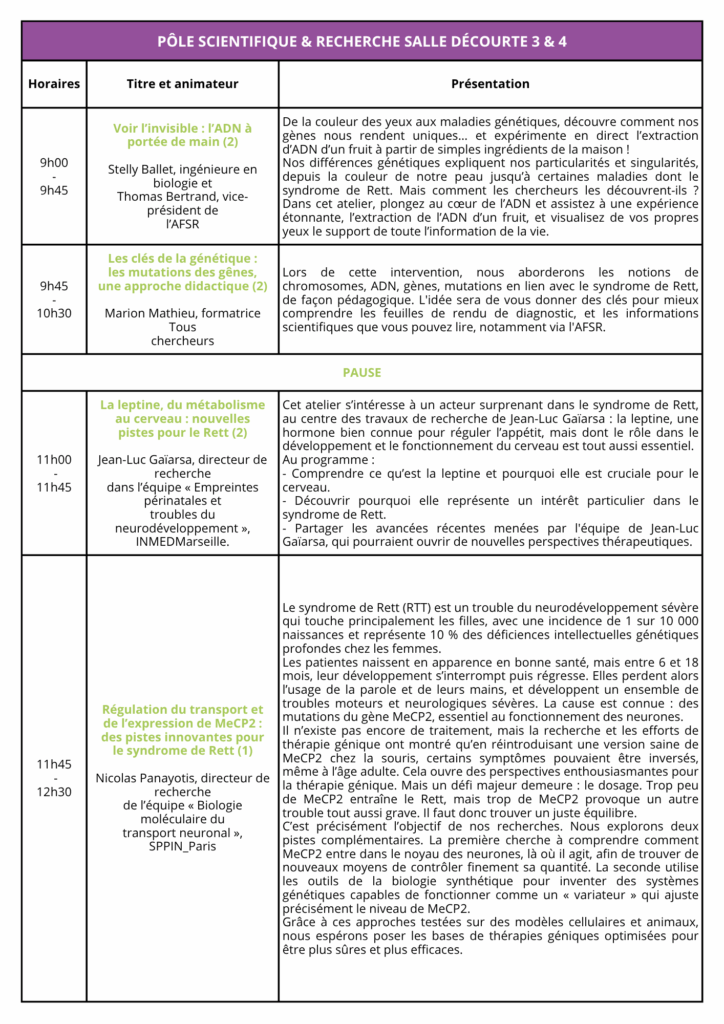
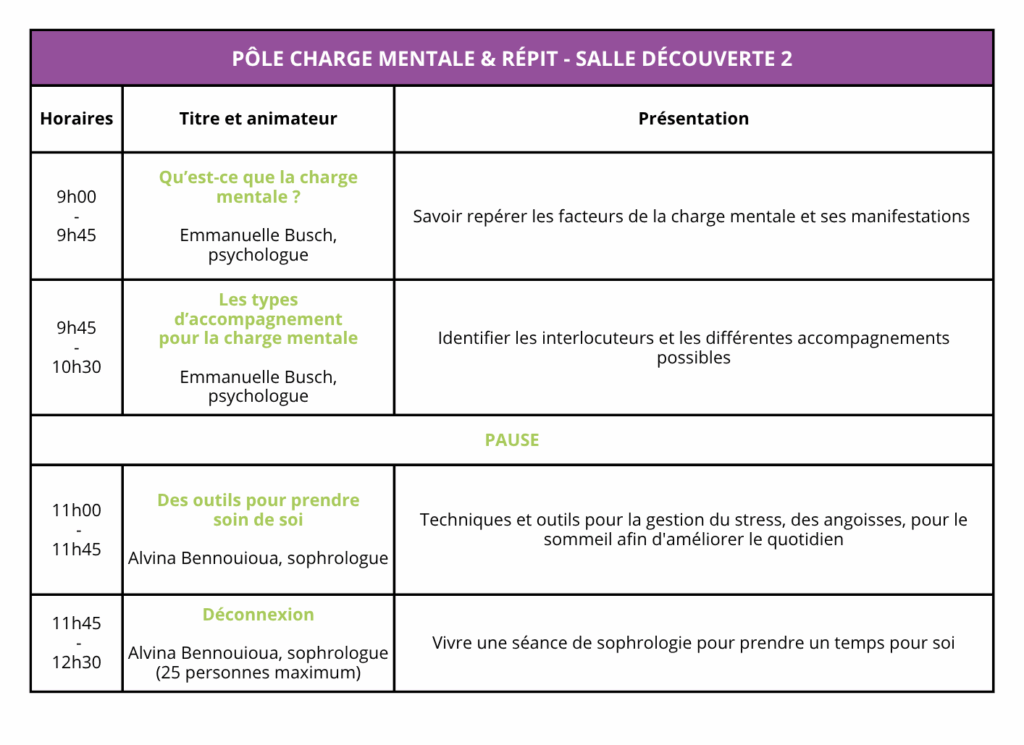
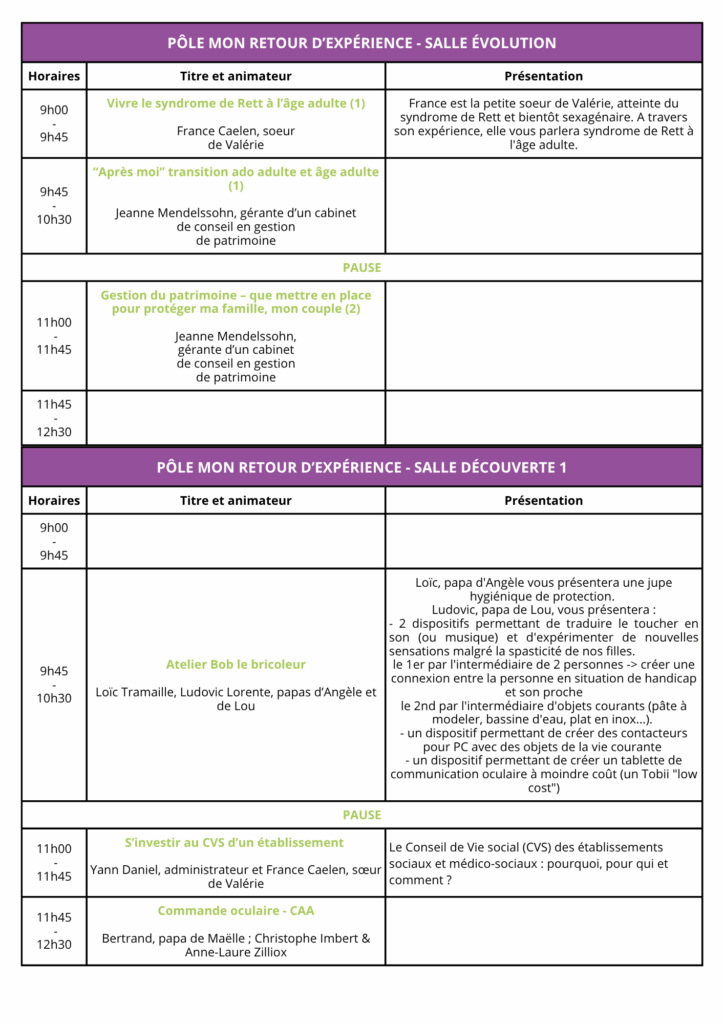
Voici une courte présentation des conférences et ateliers du dimanche.
Novotel Paris Charenton Le Pont
3-5 place des Marseillais
94227 Charenton Le Pont
Les 36èmes Journées Nationales du Syndrome de Rett de l’AFSR se sont déroulées les 12 et 13 octobre 2024 à Paris et ont accueilli plus de 130 personnes.
Cet événement dédié au syndrome de Rett, et plus largement au polyhandicap, était ouvert aux familles, mais aussi aux proches ou aidants d’une personne atteinte du syndrome de Rett, aux professionnels de santé, scientifiques, personnels de centres spécialisés, et étudiants, à tarif préférentiel.
Un programme de conférences a été proposé aux participants durant deux jours. L’Association Française du Syndrome de Rett remercie chaleureusement l’ensemble des professionnels qui ont répondu présent pour animer ces conférences.
Le programme était le suivant :

Les membres présents du Conseil d’administration et des délégations régionales se réunissaient ce samedi 12 octobre à 11h pour échanger sur les actualités de l’association et les sujets en cours. 37 membres bénévoles étaient donc présents pour l’occasion dont Séverine Agache, nouvelle déléguée régionale Hauts-de-France.
Vous pouvez retrouver par ailleurs la liste complète des membres du Conseil d’administration ici :
L’Association Française du Syndrome de Rett a accueilli ses participants à partir de 13h00 à l’espace conférence de l’hôtel Novotel.
Un badge fut remis à chaque participant. De façon traditionnelle, les nouvelles familles ont un tour de cou différent de celui des autres participants. Ceci permet à tous et toutes d’attacher une attention particulière à ces familles qui participent à l’un de nos événement pour la première fois. Elles ont d’ailleurs été accueillies lors d’un déjeuner de l’équipe du Conseil d’administration et des délégations régionales le samedi midi afin de pouvoir échanger en petit comité avant de baigner dans le grand bain !
Tous ont pu se retrouver autour d’un café d’accueil financé par Cimis.


Julien Fieschi, président, a ouvert cette 36ème édition des Journées Nationales du Syndrome de Rett par son allocution de bienvenue. Toute les séances de l’après-midi se sont tenues en réunion plénière dans l’amphithéâtre Agora. Il a présenté l’activité opérationnelle de l’année échue et les projets en cours, puis ceux restant à réaliser dans le cadre des orientations stratégiques 2020-2025. Il a également précisé qu’un travail de réflexion allait être entamé sur les orientations stratégiques de la période à venir 2025-2030.



Audrey Granado, directrice de l’AFSR, a ensuite présenté les actualités phare de l’association. Octobre Rett étant l’actualité essentielle de ce mois d’octobre a été mis en avant. Le projet de nouveau site internet a été présenté et sera mis en ligne dès la fin d’année 2024. Retrouvez les informations dans cette présentation :

Animé et modéré par Stelly Ballet, administratrice de l’Association Française du Syndrome de Rett et déléguée au Conseil Médical et Scientifique.




Julien Fieschi, président et Thomas Bertrand, vice-président de l’AFSR ont représenté la France lors du 9ème Congrès Mondial du syndrome de Rett qui s’est tenu du 2 au 5 octobre en Australie près de Brisbane. 4 jours de conférences présentées par 62 intervenants devant des participants venant de 26 pays différents. 24 associations nationales du syndrome de Rett y étaient représentées. L’occasion de faire émerger les échanges interassociatifs et de mettre en avant des actions innovantes en terme de soutien aux familles. Les scientifiques présents ont fait état de l’ensemble des projets en cours et réalisés sur la recherche fondamentale, les essais clinique et la thérapie génique. Julien Fieschi et Thomas Bertrand ont pu nous présenter l’ensemble de ces projets.


Marie-Solenne Felix, chercheuse au sein de l’équipe de Neurogénétique Humaine au centre de Génétique Médicale de Marseille (MMG) a proposé une Consulter la présentation :




Léna Bourcin, étudiante en thèse au sein de l’équipe de Neurogénétique Humaine au centre de Génétique Médicale de Marseille (MMG) a présenté le projet de thèse qu’elle porte. Cette thèse est co-dirigée par les Professeurs Jean-Christophe Roux et Jean-Luc Gaïarsa. Consulter la présentation :



Camille De Combarieu, étudiante en thèse au sein de l’équipe de Neurogénétique Humaine au centre de Génétique Médicale de Marseille (MMG) a présenté le projet de thèse qu’elle porte. Cette thèse est co-dirigée par les Professeurs Jean-Christophe Roux et Bruno Mazet. Consulter la présentation :

Rodolphe Travel, papa de Romane et adhérent de l’AFSR, récemment membre du Conseil d’administration est venu partager son expérience sur le stage Tous chercheurs proposé depuis 10 ans aux adhérents de l’AFSR grâce à l’association Tous chercheurs. Consulter la vidéo de présentation du stage :
Un stage Tous chercheurs vous sera de nouveau proposé en 2025, nous vous tiendrons informés des prochaines dates à l’avance ! Restez connectés !
Tous les participants ont pu profiter d’une pause café et jus de fruits parrainée par Cimis. Ils ont également pu profiter des temps de pause pour profiter du forum et de son espace stands avec :







Ce fut également l’occasion de pouvoir voir ou revoir l’exposition photos d’Audrey Guyon « Eléanore, ma petite sauvageonne : 10 ans avec le syndrome de Rett »





Une série de posters de portraits de bénévoles a été également exposée. L’AFSR ne vit que grâce à ses nombreux bénévoles qui se sont succédés depuis plus de 36 ans. Ensemble nous devons continuer à se relayer pour faire vivre notre communauté et porter une voix commune au nom de nos enfants et adultes. Consulter la série de posters :
Un jeu-concours a été proposé à tous les participants. En jeu, un lot de nos meilleurs articles de boutique. Nos enquêteurs devaient trouver le prénom de la jeune enfant des participants, et de la plus âgée ! Il s’agissait d’Ambre âgée de 22 mois et de Valérie, 57 ans. Une belle occasion de briser la glace et d’engager la conversation…!

Enfin, une frise participative permettait à chaque participant de s’exprimer sur :
Un grand merci pour vos contributions, celles-ci seront étudiées par le Conseil d’administration lors de l’élaboration des orientations stratégiques pour la période 2025-2030.



Sophie Tondelier, trésorière de l’AFSR animait cette seconde partie de conférences pour la journée sur le thème de répit. Après un rappel de la définition du répit et un tour d’horizon des différents modes de répit, Sophie a donné la parole à nos intervenants.



Elodie D’Andrea et Louis Dransart, fondateurs des gîtes de répit Les Bobos à la Ferme, du laboratoire du répit et bien plus que ça nous ont présenté leur projet, et nous ont expliqué pourquoi leur site est parfaitement adapté aux familles ayant un enfant atteint du syndrome de Rett. Nous saluons leurs initiatives, leur volonté d’offrir du « beau et adapté » et surtout d’accompagner les familles à monter leur projet de vacances adaptées en ayant le moins de démarches possibles à entreprendre. Consulter la présentation complète :
Laure Winterbone, référente handicap indépendante pour L’inclusion sur mesure a présenté en vidéoconférence l’ensemble des prestations pouvant être sollicitées dans le cadre du répit ainsi que leur éligibilité. Consulter la vidéo complète :
Après cet après-midi de conférences, les participants ont pu se retrouver et échanger autour d’un cocktail offert par Cimis.
Samedi soir, les échanges se sont prolongés à table jusqu’à ce qu’un groupe musical assure l’ambiance grâce à un blind-test de musique en live ! Après une journée aussi riche, nul doute que nos participants aient pu faire une bonne nuit de sommeil avant de reprendre pour le programme du dimanche !





Pascale Bridoux-Ruelle, sophrologue et maman de Léa, 30 ans, atteinte du syndrome de Rett sait à quel point il est nécessaire de prendre soin de soi et de savoir se détendre et trouver une source de relaxation dans un quotidien chargé et parfois stressant. Lors de deux ateliers, elle a proposé aux participants une séance de sophrologie.

Si vous avez participé, vous pouvez consulter son site internet et vous encourageons à laisser vos avis ici : https://maps.app.goo.gl/Yv1xniyAvN2Na6fbA
Les temps d’échange de pratique et d’expérience sont, au sein de l’association, l’une des plus grandes richesses que l’on puisse y trouver. Lors de ce week-end, nous vous avions donc proposé deux créneaux de tables rondes sur deux thématiques différentes. Le premier portait donc sur la Communication alternative améliorée (CAA) et affichait complet ! Catherine Trebaol, animatrice et médiatrice, gérait en cheffe d’orchestre ces échanges et nous a permis d’en conserver une mémoire collective. Vous pouvez la consulter ici : Synthèse de table ronde CAA.



L’AFSR ne vit que grâce à la mobilisation des familles et des donateurs. Sans cela, nous ne pourrions mettre en œuvre nos actions de soutien. Chaque année, vous êtes nombreux à nous demander comment mettre en place une manifestation de soutien. Aussi, nous avons mis en place de nombreux outils pour vous permettre d’organiser un événement « clé en main ». Audrey Granado, directrice et Eléonore Rabourdin, assistante de direction, ont présenté toutes les solutions lors de cet atelier. Consulter la présentation complète :

Les temps d’échange de pratique et d’expérience sont, au sein de l’association, l’une des plus grandes richesses que l’on puisse y trouver. Lors de ce week-end, nous vous avions donc proposé deux créneaux de tables rondes sur deux thématiques différentes. Le second portait donc sur l’adaptation du logement et affichait également complet ! Catherine Trebaol, animatrice et médiatrice, gérait en cheffe d’orchestre ces échanges et nous a permis d’en conserver une mémoire collective. Vous pouvez la consulter ici : Synthèse de table ronde aménager son logement.

Caroline Fafchamps, enseignante spécialisée et Anne-Laure Zilliox, ergothérapeute, ont proposé un atelier très divertissant ! Comment adapter le jeu avec son enfant non-verbal ? Comment adapter un moment de jeux de société en famille pour faire participer son enfant atteint du syndrome de Rett. Tout en jouant, les participants ont pu apprendre tous les trucs et astuces pour passer de bons moments en famille !



Pour ses 36èmes Journées Nationales du Syndrome de Rett, l’AFSR remercie chaleureusement l’ensemble des participants, ainsi que :
Tous ses membres bénévoles présents et engagés tout au long de l’année des délégués régionaux au Conseil d’administration ; Julien Fieschi, Thomas Bertrand ; Léna Bourcin, Camille De Combarieu et Marie-Solenne Felix « la Team Rett de Marseille » de l’équipe de recherche de l’Université d’Aix Marseille ; Stelly Ballet et Sophie Tondelier ; Elodie et Louis d’Andrea des Bobos à la ferme ; Laure Winterbone ; Caroline Fafchamps et Anne-Laure Zilliox, pour leur présence et leur implication sur nos événements et tout au long de l’année ; Eléonore Rabourdin et Audrey Granado ; Pascale Bridoux-Ruelle pour ses ateliers de sophrologie proposés bénévolement ; Catherine Trebaol ; Christophe Imbert pour sa présence sur le stand Cimis et son soutien financier pour l’organisation de ces 36èmes JNSR ; Kasia Dobrzynska pour sa présence sur le stand Acadia et leur soutien financier pour l’organisation de ces 36èmes JNSR ; Thibault Decrombecque pour sa présence et la mise à disposition gracieuse de son logiciel Eliseuse ; Audrey Guyon pour la présence et son exposition ; Handynamic pour son soutien financier Rodolphe Travel pour ses images et ses conseils précieux ; Aurélia Daniel pour la réalisation (et l’animation) des frises participatives de l’espace forum ; les fondations Borrah et Groupama pour leur soutien financier pour l’organisation de ces 36èmes JNSR.
Toute l’équipe adresse un remerciement spécial à tous les participants et aux personnes qui ont soutenu également l’AFSR en achetant ses articles de boutique.
A tous, un immense merci et rendez-vous les 11 et 12 octobre prochain pour la 37ème édition !
Les 36èmes JNSR ont été organisées avec le soutien de :




Afin de vous offrir des ateliers de qualité, nous sommes contraints de limiter le nombre de participants sur chacune des sessions proposées. Si vous souhaitez participer à un atelier le dimanche, nous vous invitons à vous y inscrire avant le 7 octobre, attention les places sont limitées !
La participation aux ateliers n’est pas obligatoire, vous pouvez aussi profiter de l’espace forum et des espaces salons de l’hôtel pour échanger avec d’autres familles.




Pour l’édition 2023, les 35èmes Journées Nationales du Syndrome de Rett de l’AFSR étaient remplacées par le 7ème Congrès Européen du Syndrome de Rett. Ces Journées se sont déroulées les 7 et 8 octobre 2023 à Marseille et ont accueilli plus de 150 personnes.
Des espoirs nouveaux en matière de recherche ont conforté les familles dans leur volonté de se regrouper autour d’une association unanimement reconnue et qui sait maintenir homogénéité et cohérence dans ses actions. Ceci vaut pour tous les pays d’Europe.
Des axes pérennes étayent la politique de nos associations comme le soutien aux familles et des efforts de toutes sortes sont menés pour grandir en représentativité et en efficacité.
Nos associations européennes ne cessent de se mobiliser pour plus qu’aucune personne atteinte par ce syndrome reste sans diagnostic. Les actions menées depuis plus de 30 ans ont pour objectif une amélioration de la qualité de vie de ces personnes.
Afin de mettre en commun, de partager nos informations, et de mutualiser nos combats, l’AFSR est honorée d’avoir organisé cette nouvelle édition à dimension européenne.
Un vaste programme de conférences a été proposé aux participants durant deux jours. L’Association Française du Syndrome de Rett remercie chaleureusement l’ensemble des professionnels qui ont répondu présent pour animer ces conférences.
Le programme était le suivant :



Nous sommes très fiers d’avoir pu réunir 150 participants venant de 24 pays d’Europe et d’ailleurs. Familles et professionnels ont partagés de chaleureux échanges tout au long du week-end.


Monsieur Julien Fieschi, président de l’Association Française du Syndrome de Rett, a accueilli les participants à cette 7ème édition du Congrès européen du Syndrome de Rett. 🇫🇷
Madame Fadila Khattabi, Ministre déléguée auprès du Ministre des Solidarités et des Familles chargée des Personnes Handicapées, a ouvert cette 7ème édition du Congrès européen du Syndrome de Rett par son discours d’ouverture. 🇫🇷
Par Anne-Sophie Lapointe, cheffe de projet de la mission maladies rares (DGOS, Paris – France) 🇫🇷


Depuis 2004 et le lancement de trois Plans nationaux successifs, la France a mis en place un dispositif unique pour accompagner les personnes malades et leur entourage. Cela est permis par le niveau d’expertise de la médecine et de la recherche en France, mais aussi par la forte mobilisation des familles, des associations et des professionnels. Cette démarche de co-construction et de co-travail a permis de poursuivre et d’amplifier la contribution des associations de malades et de leurs proches à la définition et à la mise en œuvre de la politique en faveur des maladies rares.
Objectifs pédagogiques
Le 7ème congrès Européen du Syndrome de Rett va dans ce sens. Parce que les données sont rares, l’observation clinique de la maladie rare doit servir à la recherche.
Ce qui est de l’ordre du vécu et de l’expérience du malade permet d’améliorer la connaissance sur l’histoire naturelle de la maladie.
Certaines questions ou critères importants pour la recherche peuvent être ainsi enrichis par ces données apportées par la clinique.
La clinique devient ainsi une source importante de connaissances et participe à la construction de parcours de soins mieux adaptés.
Un travail étroit avec la Banque nationale de données maladies rares (BNDMR) est régulièrement opéré dans un souci de partage des données entre le soin et la recherche, ce travail est réalisé en lien avec le système national des données de santé.
Par Pr Nadia Bahi-Buisson, neuropédiatre et responsable Centre de référence des déficiences intellectuelles de causes rares – polyhandicap (Hôpital Necker enfants malades, Paris – France) 🇫🇷


Par Rebecca Jenner, Présidente du Rett Syndrome Europe 🇪🇺

Rett Syndrome Europe, une voix forte pour les patients au niveau des médicaments et des thérapies géniques à venir.
Par Pedro Rocha, Trésorier de l’Association espagnole du Syndrome de Rett (Espagne) 🇪🇸



Par Kathie Bishop, Senior Vice-présidente, responsable de la recherche et du développement sur les maladies rares (Acadia Pharmaceuticals, San Diego – Californie) 🇺🇸
Par Edilene Siqueira, chercheuse post-doctorante (Guil’s Lab, Barcelona – Espagne) 🇪🇸

Par Jane Lunding-Larsen, chercheuse post-doctorante (Guil’s Lab, Copenhague – Danemark) 🇩🇰








Par Pr Jeffrey Neul, directeur de recherche (Centre Kennedy, Vanderbilt – Tennessee) 🇺🇸



Les progrès réalisés dans la compréhension de la physiopathologie du syndrome de Rett et le développement de modèles précliniques de la maladie ont permis de mettre au point de nouvelles approches thérapeutiques. Parallèlement, les études cliniques ont permis de préparer les essais cliniques. La combinaison de ces efforts a conduit à un avenir prometteur pour le développement de nouvelles thérapies significatives pour le syndrome de Rett.
Objectifs
Fournir une vue d’ensemble de la manière dont les informations obtenues à partir d’études de l’histoire naturelle ont permis le développement de la recherche clinique pour évaluer les nouvelles thérapies du syndrome de Rett développées par la recherche fondamentale et translationnelle.
Par Pr Mathieu Milh, Neuropédiatre et Coordinateur du Centre de référence des déficiences intellectuelles de causes rares (Hôpital La Timone, Marseille – France) 🇫🇷

Par Pr Jan-Marino Ramirez, directeur du Center for Integrative Brain Research (Seattle Children’s Research Institute, Washington – Washington) 🇺🇸


Les troubles respiratoires et autonomiques sont une caractéristique du syndrome de Rett. Cet exposé décrira les mécanismes neuronaux qui sous-tendent la dysautonomie, de la science fondamentale à la clinique.
Objectifs :
Comprendre les mécanismes qui sous-tendent la respiration erratique dans le syndrome de Rett, le rôle de l’hypoxie, le stress oxydatif et l’importance du couplage cardiorespiratoire.
Par Pr Karen Spruyt, chercheuse (INSERM NeuroDiderot, Paris – France) 🇫🇷

Chez les enfants atteints de troubles neurodéveloppementaux, les problèmes de sommeil sont plus fréquents et plus graves. Au cours de cet exposé, nous présenterons un résumé des résultats concernant le sommeil chez les personnes atteintes du syndrome de Rett.
Par Pr Anne-Marie Bisgaard, consultante (Centre Rett de l’Hôpital universitaire Rigshospitalet, Copenhague – Danemark) 🇩🇰

L’intervention comprend un examen des questions médicales et physiques et des suggestions pour le suivi des adultes atteints du syndrome de Rett, sur la base de la littérature existante et des expériences d’un centre national du syndrome de Rett.
Objectifs :









Par Pr Nicoletta Landsberger, biologiste (Université de Milan – Italie) 🇮🇹


Professeure Nicoletta Landsberger est biologiste moléculaire et a commencé à travailler sur MeCP2 aux États-Unis lorsqu’elle était post-doctorante et que MeCP2 n’avait pas encore été identifié comme la cause de la RTT. Depuis 1999, année de la découverte de la mutation de MeCP2 à l’origine de la RTT, elle a orienté ses recherches exclusivement vers le syndrome de Rett et d’autres troubles neurodéveloppementaux. En général, elle et son équipe cherchent à caractériser les altérations moléculaires et cellulaires qui sous-tendent le RTT avec pour objectif final d’identifier de nouvelles approches thérapeutiques.
Objectifs :
Par Pr Joost Gribnau, Professeur en biologie du développement (Université Erasmus, Rotterdam – Pays-Bas) NL


Par Dr Gillian Townend, Enseignante et chercheuse (Université, Reading – Royaume-Uni) 🇬🇧


Gill Townend est maître de conférences et chercheuse à l’école de psychologie et de sciences cliniques du langage de l’université de Reading au Royaume-Uni, responsable de la recherche pour Rett UK, et chercheuse indépendante au Rett Expertise Centre Netherlands. Le Dr Townend a dirigé l’équipe de projet à l’origine de l’élaboration des lignes directrices internationales en matière de communication sur le syndrome de Rett, qui ont été publiées en 2020. Elle s’intéresse également au développement de méthodologies alternatives pour l’évaluation du langage et de la cognition, à l’oculométrie et à l’utilisation fonctionnelle du regard pour la communication, ainsi qu’au traitement du langage dans le syndrome de Rett.
Par Dr Stéphane Jullien, Orthophoniste, Chargé d’enseignement, Docteur en Sciences du Langage (Centre de Logopédie de l’Université de Neuchâtel – Suisse) 🇨🇭

Professionnels et familles doivent pouvoir disposer d’une plateforme d’échange à propos du syndrome de Rett. Les professionnels doivent avoir l’occasion de se tenir au courant des avancées de la recherche et des recommandations de bonnes pratiques, notamment pour les moyens et les méthodes de communication alternatives (CAA) et les interventions orthophoniques.
L’implémentation des moyens de Communication Alternative et Améliorée (CAA) implique une collaboration transdisciplinaire entre les différents professionnels et une collaboration avec les familles à la fois pour l’évaluation comme pour les interventions. Évaluation comme intervention doivent être déployées dans le quotidien de la personne. Des grilles d’observation remplies en équipe, le partage des objectifs dans le cadre des projets individualisés sont des pratiques recommandées.
Par Meir Lotan, physiothérapeute et chercheur (Université, Ariel – Israël) 🇮🇱


Par Meir Lotan, physiothérapeute et chercheur (Université, Ariel – Israël) 🇮🇱



Par Albane Plateau, orthophoniste et chargée d’enseignement universitaire (Rouen – France) et Anne-Laure Zilliox, ergothérapeute (Luxembourg)


Par Dr Martina Semino, neuropsychomotricienne et Dr Michela, Physiotherapeute (Centre Airett, Verone – Italie) 🇮🇹


L’efficacité de la rééducation est atteinte lorsqu’elle est menée à haute fréquence et de manière généralisée ; c’est pourquoi l’AIRETT a créé un logiciel qui permet de suivre à distance la rééducation motrice (Télé-AIRETT), cognitive et communicative (AMELIE) des patients atteints du syndrome de Rett. Ainsi, ce ne sont pas les patients atteints du syndrome de Rett qui se rendent chez le spécialiste, mais le spécialiste qui se déplace à domicile, dans les écoles et les centres de rééducation des familles atteintes du syndrome de Rett.
Objectifs pédagogiques :
Faire connaître une approche de rééducation qui permet à tous les patients atteints du syndrome de Rett d’être suivis par des spécialistes ; la possibilité, à travers ce projet, de former des thérapeutes, des enseignants, des parents et de suivre la rééducation des patients atteints du syndrome de Rett. des patients atteints du syndrome de RETT
Par Laure Soulez-Larivière, diététicienne et nutritionniste (Paris – France)


Après avoir exercé le métier d’orthophoniste, je consacre maintenant mon activité à l’accompagnement diététique d’enfants et d’adultes en situation de handicap ou concernés par la maladie chronique.
Si s’alimenter est un plaisir pour la plupart d’entre nous, cela peut s’avérer difficile chez les personnes porteuses du syndrome de Rett.
Objectifs pédagogiques :
Faire le point sur les principaux troubles nutritionnels et oro-moteurs mais aussi apporter des solutions pour faciliter le temps du repas.
Par Laure Soulez-Larivière, diététicienne et nutritionniste (Paris – France)






Des nouveautés ont été présentés en exclusivité sur cette boutique tenue lors du Congrès européen : les sweats Rett dingue de Toi ont beaucoup plu ! Les achats réalisés par les participants ont permis de récolter la somme de 1 786 € ! Un grand merci à tous ! Retrouvez tous nos produits et goodies sur https://afsr.fr/boutique/.














Eléanore est née le 31 décembre 2011 et elle est atteinte du syndrome de Rett. Elle vit en Normandie avec ses parents et ses deux frères.
Le syndrome de Rett est une maladie génétique rare qui atteint le système nerveux central et qui touche principalement les filles. Il est causé par une mutation génétique à la conception. Aucun facteur héréditaire, c’est « la faute à pas de chance », comme on dit. Le syndrome de Rett, découvert en 1966, par Andreas Rett, est un polyhandicap lourd. L’épilepsie, les troubles du sommeil et de l’alimentation, la perte des acquis…font partis du tableau. Certaines filles sont marchantes, d’autres non.
Dénuée de la parole, comme la plupart des filles atteintes du syndrome, Eléanore n’en est pas moins expressive et pleine d’émotions. Avec sa maman, c’est avec la photographie qu’elles expriment l’une et l’autre leurs émotions et qu’elles renforcent le lien qui les unies.
A travers dix portraits, Audrey GUYON, maman d’Eléanore, souhaite présenter les évènements marquants de la vie de sa fille, liés au syndrome de Rett. Elle souhaite également sensibiliser le grand public au syndrome de Rett et plus largement, au handicap. Dix portraits en noir et blanc de ces dix premières années où joies et peines traversent la vie de cette petite fille extraordinaire mais où la vie et l’amour triomphent.
L’Association Française du Syndrome de Rett se joint à elle pour informer sur le syndrome. Sous chaque photographie, elle apporte des explications sur ce qui caractérise le syndrome de Rett.
Pour le 7ème Congrès européen du Syndrome de Rett, l’AFSR remercie chaleureusement l’ensemble des participants, ainsi que tous les orateurs pour leur intervention durant ce week-end. Merci à l’équipe de MCO Congrès pour leur soutien dans l’organisation de cet événement.
Toute l’équipe adresse un remerciement spécial aux bénévoles qui ont répondu présents pour nous apporter aide et soutien lors de ce week-end.
A tous, un immense merci !


Les 34èmes Journées Nationales du Syndrome de Rett de l’AFSR se sont déroulées les 12 et 13 novembre 2022 à Lille et ont accueilli près de 100 personnes.
Cet événement dédié au syndrome de Rett, et plus largement au polyhandicap, était ouvert aux familles, mais aussi aux proches ou aidants d’une personne atteinte du syndrome de Rett, aux professionnels de santé, scientifiques, personnels de centres spécialisés, et étudiants, à tarif préférentiel.
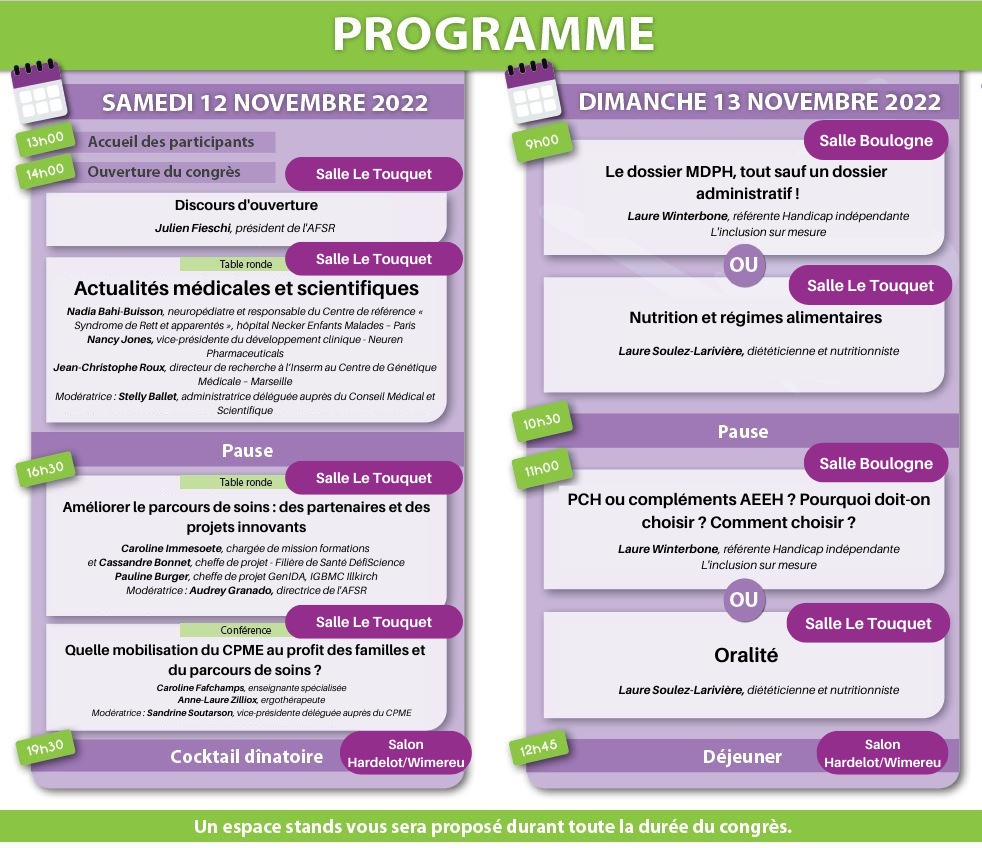
Un vaste programme de conférences a été proposé aux participants durant deux jours. L’Association Française du Syndrome de Rett remercie chaleureusement l’ensemble des professionnels qui ont répondu présent pour animer ces conférences.
Le programme était le suivant :

Le Conseil d’administration se réunissait ce samedi 12 novembre à 11h pour élire son nouveau bureau. 12 membres étaient présents pour l’occasion dont Samuel Boutillier et Hélène Jestaz, nouveaux administrateurs élus lors de la dernière assemblée générale.
Voici la constitution du nouveau bureau du Conseil d’administration :

Vous pouvez retrouver par ailleurs la liste complète des membres du Conseil d’administration ici :


L’Association Française du Syndrome de Rett a accueilli ses participants à partir de 13h00 au rez-de-chaussée de l’hôtel Mercure.
Un badge fut remis à chaque participant. De façon traditionnelle, les nouvelles familles ont un tour de cou différent de celui des autres participants. Ceci permet à tous et toutes d’attacher une attention particulière à ces familles qui participent à l’un de nos événement pour la première fois. Elles ont d’ailleurs été accueilli lors d’un déjeuner de l’équipe du Conseil d’administration et des intervenants le samedi midi afin de pouvoir échanger en petit comité avant de baigner dans le grand bain !






Julien Fieschi, président, a ouvert cette 34ème édition des Journées Nationales du Syndrome de Rett par son allocution de bienvenue. Toute les séances de l’après-midi se sont tenues en réunion plénière dans la salle Le Touquet. Il a présenté l’activité opérationnelle de l’année échue et les projets en cours, puis ceux restant à réaliser dans le cadre des orientations stratégiques 2020-2025. Consulter la présentation :

En présentiel, modéré par Stelly Ballet, administratrice de l’Association Française du Syndrome de Rett et déléguée au Conseil Médical et Scientifique.



Nancy Jones, vice-présidente du développement clinique pour Neuren Pharmaceuticals. Le laboratoire développe des nouveaux médicaments pour les patients qui souffrent de troubles du développement neurologique. Des essais cliniques en phase 3 viennent de prendre fin aux Etats-Unis sur la molécule Trofinétide. Nancy Jones nous a expliqué précisément le rôle de cette molécule et le cours des essais cliniques. Consulter la présentation :
Nadia Bahi-Buisson et Jean-Christophe Roux sont membres du Conseil Médical et Scientifique (CMS) de l’AFSR. Ils nous présenterons les actualités médicales et scientifiques dans le syndrome de Rett.






A 16h00 une pause sucrée fut servie. Des stands étaient disponibles en accès libre dans ce même espace :





En visioconférence en première partie, modéré par Audrey Granado, directrice de l’Association Française du Syndrome de Rett
Caroline Immesoete, chargée de mission formations et Cassandre Bonnet, cheffe de projet représentent toutes deux la filière de santé DéfiScience. Véritable partenaire de l’AFSR, nous développement conjointement des projets visant à améliorer le parcours des malades et celui des familles. Toutes deux parmi nous en visioconférence, Cassandre a pu présenter l’organisation de cette filière ainsi que ses missions. Caroline nous a ensuite présenté les projets en cours de réalisation et de développement.



Consulter la présentation :

Pauline Burger, cheffe de projet à l’IGBMC d’Illkirch (67) est venue nous présenter le projet GenIDA. Nous avons pu comprendre pourquoi il est indispensable d’en être acteur et de compléter de questionnaire. Consulter la présentation :



En présentiel, modéré par Sandrine Soutarson, vice-présidente de l’Association Française du Syndrome de Rett et déléguée au Conseil ParaMédical et Educatif.



Caroline Fafchamps est enseignante spécialisée en Belgique. Anne-Laure Zilliox est ergothérapeute au Luxembourg. Toutes deux sont membres du Conseil Paramédical et Educatif de l’AFSR (CPME). Elles nous ont présenté le CPME, ses missions et les membres qui la composent, ainsi que les projets réalisés, en cours et à venir. Consulter la présentation :

Enfin pour terminer cette journée, nous avons eu le plaisir de retrouver nos participants en formule 1 et 2 pour le cocktail dinatoire. Une formule plus adaptée qu’un dîner à table pour permettre de nombreux échanges entre participants !












L’inclusion sur mesure
Assistante sociale de formation initiale, j’ai travaillé dans différents secteurs avant de me spécialiser dans le domaine du handicap il y a aujourd’hui presque 15 ans. J’ai d’abord exercé en tant qu’assistante sociale avant de prendre des fonctions de direction en avant d’être Chargée de mission à la MDPH.
J’ai accompagné des professionnels et des personnes en situation de handicap adultes (SAMSAH) et enfants (SESSAD) et j’ai découvert plusieurs types de handicap : sensoriel, autisme, troubles du comportement… Mon expérience à la MDPH m’a également permis de comprendre le dossier MDPH dans toute sa globalité. Ainsi, j’ai toujours travaillé très en lien avec toutes les institutions liées au handicap.
Aujourd’hui, forte de cette expérience, je souhaite pouvoir mettre à disposition mes compétences et mon expertise au service des professionnels et des familles en tant que Référente Handicap Indépendante et au travers de la création de mon entreprise « L’inclusion sur mesure ».
Ma mission : favoriser l’inclusion.
Proposer des prestations de sensibilisation, de formation mais aussi d’accompagnement dans les parcours de soin, de scolarisation, administratif…
Ma philosophie : pragmatisme et honnêteté.
Pas de blabla mais des actions concrètes pour guider au mieux les personnes dans tout ce parcours et pour apporter des réponses adaptées à la réalité de la personne, à son environnement et à ses choix.
Je ne suis pas magicienne. Je suis juste convaincue que mieux informés, mieux accompagnés et mieux coordonnés, l’inclusion est possible !!
« Le dossier MDPH, tout sauf un dossier administratif ! »
Le dossier MDPH, passage obligé pour la reconnaissance et la compensation du handicap… mais c’est long, lourd et vécu comme une démarche administrative fastidieuse pour un résultat parfois obscur et peu compréhensible
Cet atelier vous propose de reprendre le B.A.B.A du dossier en vous expliquant les enjeux de sa bonne complétude et en vous donnant des pistes pour vous aider à le remplir correctement. Consulter la présentation :

« PCH ou compléments AEEH ? Pourquoi doit-on choisir ? Comment choisir ? »
En termes de compensation du handicap pour les enfants se pose souvent la question du choix de la PCH ou des compléments AEEH. Quelles sont les différences ? Qu’est-ce qui est le plus avantageux pour moi ?
Cet atelier vous propose d’y voir (un peu) plus clair sur ces deux prestations et leur mise en œuvre afin d’avoir des clés de compréhension pour pouvoir mieux choisir. Consulter la présentation :




Diététicienne-nutritionniste
Laure Soulez-Larivière a exercé pendant 8 ans le métier d’orthophoniste auprès de patients en situation de handicap, en particulier d’enfants souffrant de troubles alimentaires précoces associés ou non à des troubles de la déglutition. En 2016, elle se consacre au métier de diététicienne et oriente sa pratique vers les personnes de tous âges fragilisées par le handicap, la maladie chronique et la dépendance.
« Nutrition et régimes alimentaires »
La nutrition est un des piliers du bien-être. Chez l’enfant, elle permet également une croissance harmonieuse. Des conseils diététiques sont souvent très utiles pour optimiser les apports alimentaires et le confort, notamment digestif. Consulter la présentation :

« Oralité »
Si s’alimenter est un plaisir pour la plupart d’entre nous, cela peut s’avérer difficile chez les personnes porteuses du syndrome de Rett. Les troubles de l’oralité sont fréquents et une prise en charge adaptée peut considérablement aider au bon déroulement du repas. Consulter la présentation :

Pour l’occasion, retrouvez l’intégralité du reportage réalisé par Charly Fromentin sur la galerie dédiée :
Pour ses 34èmes Journées Nationales du Syndrome de Rett, l’AFSR remercie chaleureusement l’ensemble des participants, ainsi que :
Nancy Jones, Pr Nadia Bahi-Buisson, Dr Jean-Christophe Roux, Stelly Ballet, Dr Pauline Burger, Caroline Immesoete, Cassandre Bonnet, Caroline Fafchamps, Anne-Laure Zilliox, Sandrine Soutarson, Laure Soulez-Larivière ainsi que Laure Winterbone pour leur intervention durant ce week-end.
Toute l’équipe adresse un remerciement spécial aux bénévoles qui ont répondu présents pour nous apporter aide et soutien lors de ce week-end.
A tous, un immense merci !
Les 34èmes JNSR ont été organisées avec le soutien de :


L’Association Française du Syndrome de Rett remercie chaleureusement la Fondation Groupama pour son soutien.

L’Association Française du Syndrome de Rett vous accueille à partir de 13h00 au rez-de-chaussée de l’hôtel Mercure.
Un badge sera remis à chaque participant. Vous pourrez constater que les tours de cou sont de couleurs différentes. En voici le code couleur :


Julien Fieschi, président, ouvrira cette 34ème édition des Journées Nationales du Syndrome de Rett par son allocution de bienvenue. Toute les séances de l’après-midi se tiendront en réunion plénière dans la salle Le Touquet.
En présentiel, modéré par Stelly Ballet, administratrice de l’Association Française du Syndrome de Rett et déléguée au Conseil Médical et Scientifique.



Nancy Jones, vice-présidente du développement clinique pour Neuren Pharmaceuticals. Le laboratoire développe des nouveaux médicaments pour les patients qui souffrent de troubles du développement neurologique. Des essais cliniques en phase 3 viennent de prendre fin aux Etats-Unis sur la molécule Trofinétide. Nancy Jones nous expliquera précisément le rôle de cette molécule et le cours des essais cliniques.
Nadia Bahi-Buisson et Jean-Christophe Roux sont membres du Conseil Médical et Scientifique (CMS) de l’AFSR. Ils nous présenterons les actualités médicales et scientifiques dans le syndrome de Rett.
Une pause sucrée vous sera servie à l’espace Patio. Des stands seront disponibles en accès libre dans ce même espace :
Ces stands seront également disponibles à d’autres moment du congrès. N’hésitez pas à aller à leur rencontre !
Les stands seront spécifiquement assurés dans ces créneaux horaires :
Samedi de 13h00 à 14h00, de 16h00 à 16h30 et de 18h00 à 19h30
Dimanche de 10h30 à 11h00
En visioconférence en première partie, modéré par Audrey Granado, directrice de l’Association Française du Syndrome de Rett
Caroline Immesoete, chargée de mission formations et Cassandre Bonnet, cheffe de projet représentent toutes deux la filière de santé DéfiScience. Véritable partenaire de l’AFSR, nous développement conjointement des projets visant à améliorer le parcours des malades et celui des familles. Elles seront parmi nous en visioconférence pour vous présenter les projets en cours de réalisation et de développement.
Pauline Burger, cheffe de projet à l’IGBMC d’Illkirch (67) vous présentera le projet GenIDA et vous expliquera pourquoi il est indispensable d’en être acteur et de compléter de questionnaire.
En présentiel, modéré par Sandrine Soutarson, vice-présidente de l’Association Française du Syndrome de Rett et déléguée au Conseil ParaMédical et Educatif.
Caroline Fafchamps est enseignante spécialisée en Belgique. Anne-Laure Zilliox est ergothérapeute au Luxembourg. Toutes deux sont membres du Conseil Paramédical et Educatif de l’AFSR (CPME). Elles vous présenteront le CPME, ses missions et les membres qui la composent, ainsi que les projets réalisés, en cours et à venir.
La fin des conférences est prévue vers 18h. Vous pourrez profiter de votre temps libre avant de nous retrouver pour le cocktail dinatoire.
Dès 19h, nous avons le plaisir de vous retrouver, participants en formule 1 et 2, pour le cocktail dinatoire qui sera servi en salle Hardelot/Wimereu.

L’inclusion sur mesure
Assistante sociale de formation initiale, j’ai travaillé dans différents secteurs avant de me spécialiser dans le domaine du handicap il y a aujourd’hui presque 15 ans.
J’ai d’abord exercé en tant qu’assistante sociale avant de prendre des fonctions de direction en avant d’être Chargée de mission à la MDPH.
J’ai accompagné des professionnels et des personnes en situation de handicap adultes (SAMSAH) et enfants (SESSAD) et j’ai découvert plusieurs types de handicap : sensoriel, autisme, troubles du comportement… Mon expérience à la MDPH m’a également permis de comprendre le dossier MDPH dans toute sa globalité. Ainsi, j’ai toujours travaillé très en lien avec toutes les institutions liées au handicap.
Aujourd’hui, forte de cette expérience, je souhaite pouvoir mettre à disposition mes compétences et mon expertise au service des professionnels et des familles en tant que Référente Handicap Indépendante et au travers de la création de mon entreprise « L’inclusion sur mesure »
Ma mission : favoriser l’inclusion.
Proposer des prestations de sensibilisation, de formation mais aussi d’accompagnement dans les parcours de soin, de scolarisation, administratif…
Ma philosophie : pragmatisme et honnêteté.
Pas de blabla mais des actions concrètes pour guider au mieux les personnes dans tout ce parcours et pour apporter des réponses adaptées à la réalité de la personne, à son environnement et à ses choix.
Je ne suis pas magicienne.
Je suis juste convaincue que mieux informés, mieux accompagnés et mieux coordonnés, l’inclusion est possible !!
« Le dossier MDPH, tout sauf un dossier administratif ! »
Le dossier MDPH, passage obligé pour la reconnaissance et la compensation du handicap… mais c’est long, lourd et vécu comme une démarche administrative fastidieuse pour un résultat parfois obscur et peu compréhensible
Cet atelier vous propose de reprendre le B.A.B.A du dossier en vous expliquant les enjeux de sa bonne complétude et en vous donnant des pistes pour vous aider à le remplir correctement.
« PCH ou compléments AEEH ? Pourquoi doit-on choisir ? Comment choisir ? »
En termes de compensation du handicap pour les enfants se pose souvent la question du choix de la PCH ou des compléments AEEH. Quelles sont les différences ? Qu’est-ce qui est le plus avantageux pour moi ?
Cet atelier vous propose d’y voir (un peu) plus clair sur ces deux prestations et leur mise en œuvre afin d’avoir des clés de compréhension pour pouvoir mieux choisir.

Diététicienne-nutritionniste
Laure Soulez-Larivière a exercé pendant 8 ans le métier d’orthophoniste auprès de patients en situation de handicap, en particulier d’enfants souffrant de troubles alimentaires précoces associés ou non à des troubles de la déglutition. En 2016, elle se consacre au métier de diététicienne et oriente sa pratique vers les personnes de tous âges fragilisées par le handicap, la maladie chronique et la dépendance.
« Nutrition et régimes alimentaires »
La nutrition est un des piliers du bien-être. Chez l’enfant, elle permet également une croissance harmonieuse. Des conseils diététiques sont souvent très utiles pour optimiser les apports alimentaires et le confort, notamment digestif.
« Oralité »
Si s’alimenter est un plaisir pour la plupart d’entre nous, cela peut s’avérer difficile chez les personnes porteuses du syndrome de Rett. Les troubles de l’oralité sont fréquents et une prise en charge adaptée peut considérablement aider au bon déroulement du repas.
Une pause salée sera servie dimanche de 10h30 à 11h à l’espace Le Patio. L’occasion d’aller à la rencontre des stands et repartir avec un article de notre boutique !
Dès 12h45, nous avons le plaisir de vous retrouver, participants en formule 1 et 2, pour le déjeuner qui sera servi en salle Hardelot/Wimereu. Au menu :
Plat : Filet de volaille, fricassé de légumes et crème de poule au pot
Dessert : Moelleux aux pommes tiède, glace spéculoos et caramel au beurre salé
Eaux minérales et thé/café inclus
Nous vous remercions sincèrement pour votre participation ! Un questionnaire de satisfaction vous sera envoyé directement par mail, nous comptons sur vous pour le compléter.
Toute l’équipe vous souhaite un bon retour !
Les 33èmes Journées Nationales du Syndrome de Rett de l’AFSR se sont déroulées les 6 et 7 novembre 2021 à Lyon et ont accueilli plus de 140 personnes.
Cet événement dédié au syndrome de Rett, et plus largement au polyhandicap, était ouvert aux familles, mais aussi aux proches ou aidants d’une personne atteinte du syndrome de Rett, aux professionnels de santé, scientifiques, personnels de centres spécialisés, et étudiants, à tarif préférentiel.
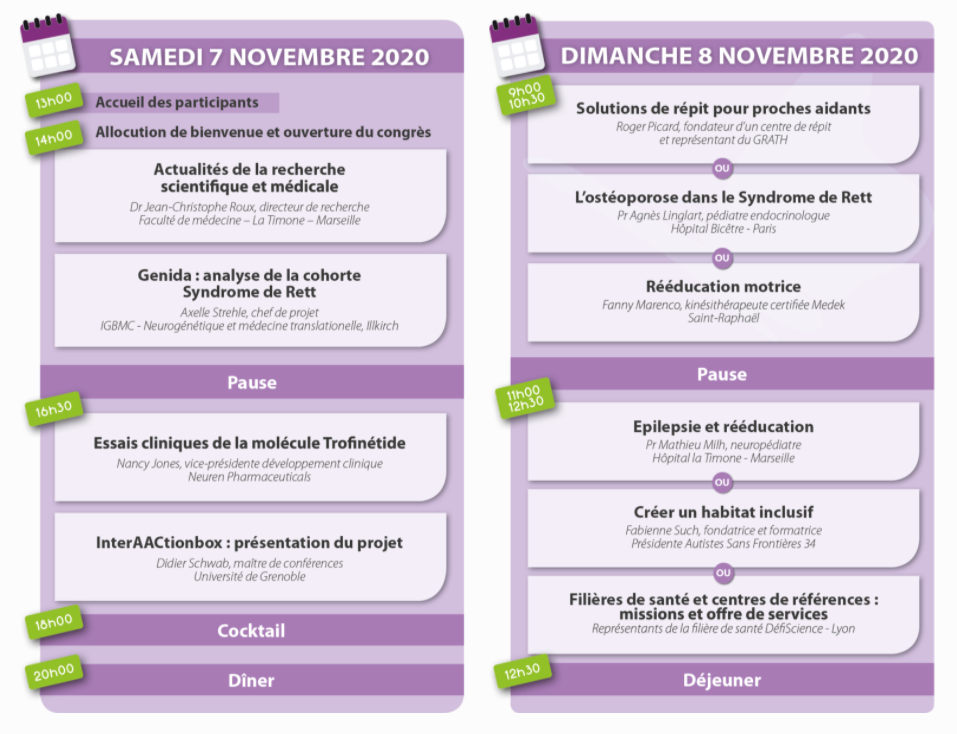
Un vaste programme de conférences a été proposé aux participants durant deux jours. L’Association Française du Syndrome de Rett remercie chaleureusement l’ensemble des professionnels qui ont répondu présent pour animer ces conférences.
Le programme était le suivant :


Julien Fieschi, président de l’AFSR
Vous retrouverez ici l’allocution de bienvenue de Marie Garnier :


Audrey Granado, directrice de l’AFSR

Dr Jean-Christophe Roux, directeur de recherche – Faculté de médecine – La Timone
Dr Jean-Christophe Roux – Faculté de médecine – Marseille

Didier Schwab, maître de conférences – Université de Grenoble

Marie Voisin Du Buit, orthophoniste
L’apraxie :
∙ L’apraxie : est l’un des symptômes très présent et le plus handicapant dans le syndrome de Rett
⇨ Sous-jacent dans pleins de problématique dans le syndrome de Rett
∙ Apraxie : Incapacité d’effectuer des mouvements moteurs fonctionnels en dépit de :
● La compréhension de la consigne
● La volonté de faire le mouvement
● Les aptitudes physiques (force musculaire, etc.)
⇨ L’apraxie est d’origine neurologique
∙ Exemple :
Quand on demande à un enfant de montrer le chien parmi des images, la difficulté ne va pas être de
comprendre ce qu’on lui demande (consigne : montre-moi le chien), car l’enfant est capable de
reconnaitre l’image (il connait et sait ce qu’est le chien). Mais la difficulté est d’effectuer le mouvement
pour arriver à le montrer (c’est ça qui pose problème).
Ça peut prendre beaucoup de temps pour aller de l’initiation/intention à l’action et c’est ce qui explique
ces délais de réponses (on observe que le délai de réponses est extrêmement long chez les personnes
atteintes du syndrome de Rett)
Impact sur les compétences socles du langage oral :
∙ Les gestes à visée communicative (pointage, etc) sont souvent absents
∙ Le suivi oculaire dirigé ou coordonné pour permettre l’attention conjointe est parfois difficile à
maintenir, même s’il est considéré comme un point fort
∙ Les signaux non-verbaux (mimiques, etc) sont limités
Conséquences directes :
∙ Les compétences de la personne sont sous-estimées (compétences cognitives et communication)
∙ On ne propose pas d’outil suffisant et adapté
∙ Facteur de découragement et de perte d’estime de soi
Que faire ?
⇨ Faut-il rééduquer l’apraxie ? travailler le pointage, les gestes ?
Évaluation mauvaise si l’enfant ne connait pas de prime abord son interlocuteur
● Il faut vraiment apprendre à connaitre l’enfant avant
● Savoir ces moyens de communications
● Bien faire attention entre consignes directes et indirectes

Estelle Richez, orthophoniste et Cécile Sabatier, éducatrice spécialisée à domicile
La modélisation c’est quoi ?
∙ Utiliser l’outil de CAA = combiner son utilisation et le langage oral
∙ Par exemple : pointer les pictogrammes et parler ou signer et parler
∙ Attention modéliser ce n’est pas calquer un discours, parler à la place de personne ou imiter
∙ C’est parler avec ses mots pour créer des situations de communication et non d’imitation
Pourquoi modéliser ?
∙ Pour apprendre à parler une langue il est crucial d’être entouré de personnes qui parlent elles-mêmes cette langue c’est « le bain de langage »
∙ La CAA doit aussi s’inscrire dans cette pratique écologique et quotidienne
∙ La CAA ça s’apprend, il n’y a pas de prérequis nécessaire à l’introduction d’un outil de CAA
● Il faut commencer par proposer l’outil et l’introduire dans le quotidien afin de permettre à la
personne de s’en saisir
∙ Comment demander à quelqu’un d’utiliser un outil si on ne lui a jamais permis d’apprendre
∙ Fonctions de communication ; le langage rempli différentes fonctions
∙ Souvent on a tendance à se concentrer sur une ou deux fonctions (la demande par exemple)
∙ Mais communiquer c’est bien plus que simplement demander
Comment modéliser ?
∙ L’importance des petits pas
∙ En CAA pour avancer sans se décourager mieux vaut se donner des petits objectifs
∙ Les réévaluer régulièrement en fonction de l’évolution des capacités des envies et des besoins
∙ Se fixer des petits objectifs :
● En respectant la zone proximale de développement = ajuster notre comportement en fonction
des capacités de la personne et lui proposer d’aller juste un peu plus loin
● En modélisant du vocabulaire de base et du vocabulaire spécifique
● En modélisant différentes natures de mots (noms, verbes, petits mots, adjectifs)
∙ En proposant la multimodalité (signes, pictogrammes)
∙ En utilisant différents supports en fonction des besoins et des contraintes (outils robustes mais aussi
outils de base)
● Exemple : un TLA (Tableau de langage assisté) pour le bain, là où la tablette ne peut pas aller
⇨ Réévaluer ces objectifs régulièrement en fonction de l’évolution des capacités des envies et des
besoins
∙ Concrètement :
● Se lancer et déculpabiliser
● Pratiquer
● Donner l’accès aux outils (disponible en état de marche)
● Donner du temps, être patient, persévérant
● Créée des opportunités / saisir les opportunités
● Être authentique
● Croire en son potentiel
La modélisation comment commencer ?
Exemples tirés du quotidien
∙ Quel moment de la journée pouvez-vous consacrer à la communication avec votre enfant ?
● Un moment ou vous êtes disponible ou vous ne serez pas dérangé
● Un moment ou votre enfant est reposé et disponible
● Un moment ou vous pourrez vous rendre disponible dans la régularité
∙ Qu’est ce qui peut intéresser votre enfant ?
∙ Avez-vous remarqué chez votre enfant un ou plusieurs centres d’intérêts ?
∙ Que se passe-il autour de ce moment
∙ Qu’est ce qui intéresse votre enfant dans l’activité
∙ Comment réagit-il ?
∙ Veut-il recommencer, arrêter ?
⇨ Petit à petit de plus en plus de possibilités
∙ Quelques points importants :
● Utiliser toujours la même image pour le même mot
● Encourager les initiatives de vote enfant
● Accepter d’aller à son rythme
● Lui laisser le temps de prendre la parole de compléter ou de répondre
∙ Viser haut, mais commencer petit

Caroline Fafchamps, enseignante spécialisée en Belgique
L’odyssée de Julie et Pablo – L’odysée de Julie et Pablo (odyseedejulieetpablo.be)
L’odyssée De Julie et Pablo – Accueil | Facebook
https://www.facebook.com/groups/415787173128071

Orane Chauvin, ergothérapeute

Anne-Laure Zilliox, ergothérapeute
Présentation et explication de la méthode Cuevas MEDEK Exercices
Fanny Marenco, kinésithérapeute – Nice
Voici le lien pour trouver un praticien utilisant la méthode CME-MEDEK en France :
https://www.google.com/maps/d/viewer?mid=1oJtQMksrpNrNG5e5ql55u7-QuL4&usp=sharing

Léa Lebzar, ergothérapeute – Lunel
La mauvaise intégration de ces réflexes archaïques peut entrainer des difficultés au niveau du développement psychomoteur, de ses capacités d’apprentissage et de communication, ainsi que de son comportement.
C’est pourquoi il est important de faire émerger et d’intégrer ces nombreux réflexes.
3. Intégration des réflexes archaïques selon la méthode Rmti (Rhythmic Movement Training International)
Les objectifs seront de :
Le programme comportera :
auxquels sera toujours associé un aspect ludique
Comment trouver un professionnel Rmti ?
Les familles intéressées par la méthode et qui souhaitent trouver un thérapeute dans leur région, peuvent contacter Léa ( lebzar.lea@gmail.com ) ou consulter l’annuaire des personnes ayant une certification Rmti : https://www.rhythmicmovement.org/consultants
Jeanne Mendelssohn, conseil en gestion sociale et patrimoniale – Paris

Me Baptiste Canonville, avocat en droit du handicap – Nantes
Lors des 33èmes JNSR, des stands ont permis d’accueillir :
Pour ses 33èmes Journées Nationales du Syndrome de Rett, l’AFSR remercie chaleureusement l’ensemble des participants, ainsi que :
Dr Jean-Christophe Roux, Didier Schwab, Marie Voisin Du Buit, Estelle Richez, Caroline Fafchamps, Orane Chavin, Anne-Laure Zilliox, Fanny Marenco, Léa Lebzar, Jeanne Mendelssohn ainsi que Me Baptiste Canonville pour leurs interventions durant ce week-end.
Toute l’équipe adresse un remerciement spécial aux bénévoles qui ont répondu présents pour nous apporter aide et soutien lors de ce week-end ainsi qu’à l’élaboration des comptes-rendus.
A tous, un immense merci !
Les 33èmes JNSR ont été organisées avec le soutien de :

Malheureusement, en raison de l’actualité sanitaire, nous n’avons pas pu maintenir notre édition des JNSR les 7 et 8 novembre à Lyon. Vous trouverez tout de même ici quelques supports fournis par nos professionnels qui avaient accepté d’intervenir pour l’occasion. Cette liste sera actualisée à mesure que les professionnels nous transmettrons leur support. Nous vous en tiendrons informés.
A l’origine, nous avions envisagé d’accueillir Pauline Burger pour qu’elle nous présente l’analyse des résultats obtenus relatifs à la cohorte de patients atteints du syndrome de Rett. Nous avions consacré au projet une page du dernier Rett info. Malheureusement, trop peu de réponses ont été apportées pour pouvoir en tirer une quelconque analyse. Nous tenterons alors ici de vous convaincre d’être vous aussi acteur de ce projet participatif.
Pauline Burger, Cheffe de projet GenIDA
Le projet GenIDA en quelques mots
Le projet GenIDA est une étude participative initiée en 2016 par le Pr Jean-Louis Mandel, Université de Strasbourg, ayant pour objectif de mieux caractériser les manifestations cliniques et histoires naturelles des formes génétiques de troubles neurodéveloppementaux.
Le site internet https://genida.unistra.fr permet aux familles de s’inscrire et de répondre à un questionnaire structuré constitué principalement de “questions à choix multiple” (compter 1 h pour le remplir en moyenne). Ces questions explorent notamment les paramètres physiques (taille, poids), les aspects cognitifs (compétences pour le langage parlé, la lecture et l’écriture, communication non verbale), les aspects comportementaux, la présence ou non de manifestations cliniques neurologiques (épilepsie, troubles moteurs) ou affectant les grandes fonctions physiologiques (cardiaque, rénale, gastro-intestinale, sensorielles, comportement alimentaire, etc.).
Cinq questions ouvertes explorent de manière plus qualitative la perception par les familles de manifestations qui affectent le plus la santé de leur proche, la qualité de vie, l’existence éventuelle de réactions indésirables à des traitements ciblés ou symptomatiques. Ces questions permettent aux familles de mettre en avant des aspects de la maladie qui leur apparaissent comme majeurs.
Les données recueillies dans le cadre de cette étude sont analysées statistiquement de manière anonyme, et des visualisations des résultats sont possibles dès que la taille de la cohorte le permet.
Cette approche est inhabituelle de par l’implication directe des familles. Notre expérience sur les syndromes Koolen-deVries, Kleefstra et KBG démontre que les parents sont véritablement “experts”. Ils peuvent non seulement parfaitement synthétiser les manifestations associées à la pathologie (comme le feraient des professionnels), décrire leurs effets sur la qualité de vie ou l’autonomie de leurs enfants, mais surtout révéler des aspects de la pathologie jusqu’alors sous-estimés. De plus, les parents sont en général particulièrement motivés pour faire progresser les connaissances (comme en témoigne l’essor impressionnant des associations de patients et familles pour les maladies rares, sur les réseaux sociaux), et sont ceux qui sont le plus avides de réponses à leurs questionnements sur l’avenir de l’enfant.
Nous espérons que ce projet permettra d’améliorer les connaissances sur de nombreuses formes de déficience intellectuelle, en apportant des données nouvelles et utiles aux patients et familles concernées, et aux professionnels (médecins mais aussi psychologues et éducateurs) qui suivent les personnes atteintes.
Le projet GenIDA est financé par la Fondation de l’Université de Strasbourg (Fonds Roche pour la médecine personnalisée), par l’USIAS (University of Strasbourg Institute for Advanced Studies), la Fondation Jérôme Lejeune et le GIS-Autisme. Le projet a bénéficié de l’aide et des conseils du programme national RADICO pour les aspects légaux et éthiques (dossiers CNIL n°1907912 et comité d’éthique de l’INSERM n°16-338) et de sécurité informatique.
Accroitre la taille de la cohorte participative est essentiel pour faire émerger des nouvelles informations d’intérêt médical, permettant toujours une meilleure prise en charge des personnes atteintes.
Pour cela, rendez-vous sur le site https://genida.unistra.fr, inscrivez-vous, approuvez le formulaire de consentement et répondez au questionnaire général à propos de votre proche après avoir activé son profil.
A noter :
Une vidéo (en anglais mais très didactique) est disponible sur notre chaine Youtube pour vous guidez dans ces démarches : https://youtu.be/-8eJD9Chbe4
Si toutefois vous aviez des questions concernant le projet GenIDA ou si vous rencontriez des problèmes lors de votre inscription sur le site, n’hésitez pas à nous contacter :
Par mail : burgerp@igbmc.fr ; genida@igbmc.fr
Via notre page Facebook (Messenger) : www.facebook.com/GenIDAproject/
Aidez-nous à vous aider :
L’équipe GenIDA
Pauline BURGER et Jean-Louis MANDEL
burgerp@igbmc.fr ; genida@igbmc.fr
Tel: +33 (0)3 88 65 56 25
Website: genida.unistra.fr/
Facebook: www.facebook.com/GenIDAproject/
YouTube Channel « Genida Project »: https://www.youtube.com/channel/UCHR_4upMiE33Q4tcxp6kJ_Q
Nancy Jones, Vice-présidente développement clinique de Neuren Pharmaceuticals
Neuren Pharmaceuticals développe des nouveaux médicaments pour les patients qui souffrent de troubles du développement neurologique. Des essais cliniques en phase 3 sont en cours aux Etats-Unis sur la molécule Trofinétide. Nancy Jones, vice-présidente du développement clinique, qui aurait du être également présente lors de notre congrès annuel, a enregistré pour nous une vidéo dans laquelle elle nous explique précisément le rôle de cette molécule et le cours des essais cliniques.
Nous la remercions sincèrement pour cette vidéo et suivons de près la suite de ces essais et la publication des résultats et vous tiendrons bien entendu informés.
Didier Schwab, Maître de conférences – Université de Grenoble-Alpes
L’InterAACtionBox soutenu par un financement de l’Association Française du Syndrome de Rett vise à aider les personnes en situation de handicap cognitif. Cet handicap les empêche de communiquer par la parole et de se développer intellectuellement via cette unique modalité langagière. Il s’agit de concevoir un dispositif informatique à moindre coût, proposant des applications au plus près de la recherche et des utilisateurs, ouvert, adaptable et évolutif qui vont permettre l’éveil, le jeux, l’éducation mais aussi le développement communicatif et langagier des enfants avec un polyhandicap.
Le projet se positionne au carrefour de la cognition, de l’interaction homme-machine et du traitement automatique de la langue et de la parole. Il fédère les forces de chercheurs issus de plusieurs laboratoires de l’Université Grenoble Alpes, informaticiens, traducteurs, linguistes et psycholinguistes, dont certains sont aussi parents d’enfants en situation de polyhandicap. Le projet est aussi développé en collaboration avec des professionnels du terrain en orthophonie, ergothérapie, orthoptie et neuropsychologie ainsi que des personnes proches du quotidien des personnes concernées (éducateurs et autres professionnels intervenant en institution et des aidants).
Cet appareil intègre un écran tactile et est compatible avec des appareils oculométriques, précis, robustes, et peu onéreux, pour un prix avoisinant les 500 €.
Sur le plan logiciel, il s’agit de concevoir et d’implémenter une batterie de logiciels libres et ouverts. Ces logiciels sont actuellement développés au Laboratoire d’Informatique de Grenoble, en collaboration avec des membres du Gipsa-Lab en interaction avec des étudiants de différents cursus et avec des objectifs d’apprentissage, de divertissement mais aussi d’évaluation. On y retrouve :
– GazePlay, une plateforme de jeux utilisables par oculométrie en développement depuis 2016 et en constante évolution proposant à ce jour plus de 60 jeux créatifs, ludiques ou sérieux permettant d’aider à l’acquisition de compétences (action-réaction, sélection, littératie, mémorisation, etc.). Sur le long terme, GazePlay comportera aussi une partie évaluation (GazePlay-Eval) qui fait l’objet de projets de mémoire en neuropsychologie ;
– VisualSceneDisPlay, un logiciel interactif et configurable de scènes visuelles pour apprendre le vocabulaire de base aux enfants, tout en faisant un premier pas vers la Communication Alternative et Augmentée ;
– Augcom, un outil de grille de Communication Alternative et Augmenté, basé sur des études comparatives des différents logiciels existants, sur les besoins exprimés par les familles et qui continuera à s’améliorer en proposant notamment un lexique et une organisation optimales basés sur la recherche ;
– L’InterAACtionBox assurera également un accès aux sites de streaming musicaux préférés des utilisateurs, tels que Youtube ou deezer.
Une série de webinaires et un stage devraient être organisés par l’AFSR dès 2021 sur le sujet. Nous négocions actuellement des tarifs afin de vous proposer dans le courant de l’année l’InterAACtionbox-AFSR clé en main à un tarif accessible à tous. Nous ne manquerons pas de vous informer par mail, mais vous pouvez également compléter le formulaire qui se trouve en page d’accueil du site dédié.
Plus d’infos : https://interaactionbox.afsr.fr/
Caroline Immesoete, chargée de mission pôle formation
L’AFSR dépend de plusieurs filières de santé dont DéfiScience, dont dépend notamment le « Centre Rett et apparentés » dirigé par le Pr Bahi-Buisson à Paris et celui du Pr Mathieu Milh à Marseille. La filière est à l’origine du PNDS Syndrome de Rett et apparentés mais également du PNDS Polyhandicap paru récemment. Vous trouverez ci-dessus le lien vers le document de présentation de la filière fourni par Madame Immesoete.
Des liens étroits se tissent entre les filières de Santé et les associations de familles et de patients. Des projets sont également en cours et naitront de ces partenariats dans les mois à venir. Nous ne manquerons pas de vous tenir informés.

Accéder au document de présentation fourni par la filière de santé au format PDF
Les inscriptions seront prochainement ouvertes.

















