



Les 31èmes Journées Nationales du Syndrome de Rett de l’AFSR se sont déroulées les 5 et 6 octobre 2019 à l’hôtel Ibis Paris 17 Clichy-Batignolles et ont accueilli plus de 240 personnes.
Cet événement dédié au syndrome de Rett, et plus largement au polyhandicap, était ouvert aux familles, mais aussi pour la première fois, aux proches ou aidants d’une personne atteinte du syndrome de Rett, aux professionnels de santé, scientifiques, personnels de centres spécialisés, et étudiants, à tarif préférentiel.
Un vaste programme de conférences a été proposé aux participants durant deux jours. L’Association Française du Syndrome de Rett remercie chaleureusement l’ensemble des professionnels qui ont répondu présent pour animer ces conférences.
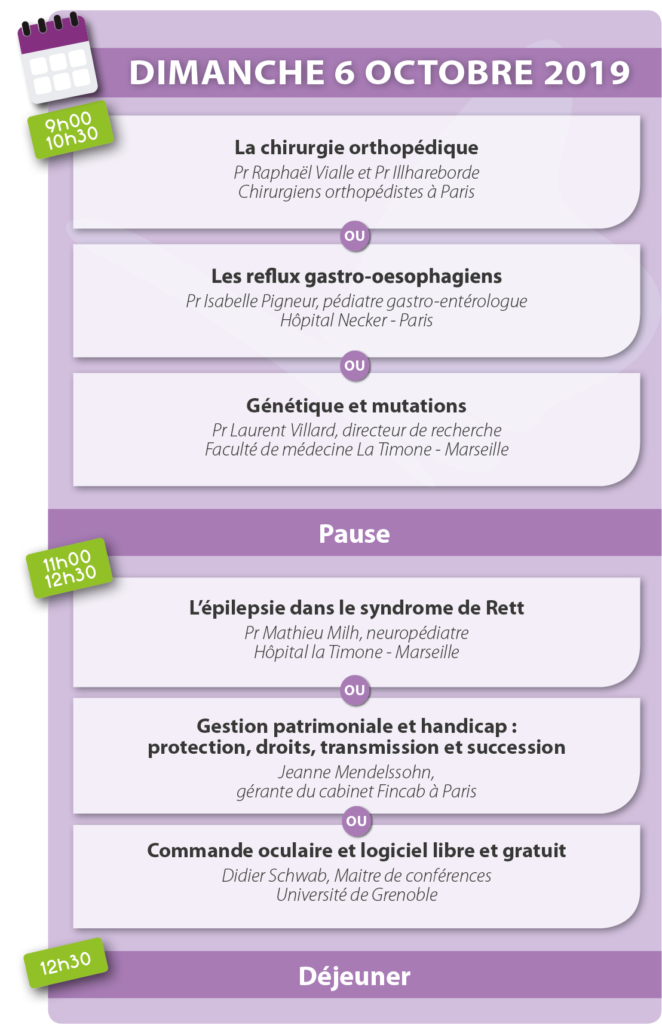
Le programme était le suivant :

 Accéder au diaporama présenté lors de cette conférence
Accéder au diaporama présenté lors de cette conférence
Julien Fieschi se présente ainsi que le conseil d’administration (15 membres).
Il continue en présentant les 4 axes prioritaires de l’association pour la période 2020-2025 (https://afsr.fr/orientations-2020-2025/) :
Ces 4 axes prioritaires vont guider l’action de l’association durant les 5 prochaines années.
Il annonce également qu’une grande consultation sera lancée en novembre vers tous les adhérents de l’AFSR pour recueillir leurs avis et opinions sur différents sujets.
Il présente brièvement les 23 filières de santé maladie rares issues du 3ème plan ‘Maladies rares’ du ministère de la santé (2018-2020). Le syndrome de Rett est inclus dans plusieurs filières dont la principale est ‘Défiscience’ (https://www.defiscience.fr/). Il rappelle qu’il existe en France 27 centres de référence et 79 centres de compétences. Il est intéressant d’utiliser les centres de compétence de nos régions, non seulement pour avoir un suivi de proximité (notamment en cas d’urgence) mais également pour que les connaissances des professionnels de santé sur le syndrome de Rett se développent aussi dans ces centres (et ne restent pas seulement concentrées sur quelques endroits du territoire).
Il décline enfin la stratégie de recherche de l’AFSR pour la période 2020-2025. Jusqu’à maintenant l’AFSR a soutenu un total de 62 projets de recherche pour un montant total de 1 200 000 euros. L’AFSR va dorénavant articuler ses projets de recherches autour de deux axes clairs:
A cette occasion, il annonce le lancement durant le mois d’octobre qui est le mois de sensibilisation du Syndrome de Rett, d’un appel à dons via les adhérents de l’association pour financer un appel à projet d’un montant de 200 000 euros pour travailler sur des thérapies innovantes (génétique ou cellulaire).
 Accéder au diaporama présenté lors de cette conférence
Accéder au diaporama présenté lors de cette conférence
Membre du Conseil Médical et Scientifique de l’AFSR, le Dr Jean-Christophe Roux est directeur de recherche à l’INSERM de Marseille au sein de l’équipe Neurogénétique Humaine, dont les deux thèmes d’études principaux sont le syndrome de Rett et les encéphalopathies épileptiques précoces.
La recherche médicale et scientifique et les essais cliniques suscitent toujours beaucoup d’intérêt au sein de l’association. La thérapie génique est un des sujets les plus sollicités par les parents.
Les espoirs sont grands pour les familles, mais que peut-on vraiment espérer ? Où en est-on ?
Jean-Christophe Roux nous a présenté un résumé à la fois des difficultés et de la complexité de la thérapie génique dans le syndrome de Rett ainsi que les avancées et les espoirs.
On peut rappeler qu’il existe deux grands types de recherche thérapeutique, la pharmacologie (largement dominante) qui a une action à court terme et traite les symptômes et les thérapies de substitution (thérapie géniques, réactivation du chromosome X et passage forcé du codon stop) qui visent à traiter les causes (« corriger » les mutations génétiques dans notre cas).
Parmi ces dernières on peut distinguer :
1-thérapie de passage forcé du codon stop (pour certains types de mutation uniquement – mais représentant 50% des patients ayant Rett)
2-la réactivation du chromosome X (un seul X étant activé dans chaque cellule la réactivation du second chromosome X ne possédant pas la mutation dans les cellules malades devrait guérir ces cellules)
3-thérapies génique visant à introduire un gène fonctionnel à l’aide d’un vecteur viral ou non-viral.
Définition de la thérapie génique : introduire des acides aminés (blocs de base du code génétique, ADN) dans les cellules pour altérer l’évolution d’une condition médicale. A l’heure actuelle, 60% des essais de thérapie génique sont développés dans le contexte de la recherche sur le traitement des cancers.
Le syndrome de Rett est un bon candidat pour la thérapie génique puisqu’il est mono-génétique et que sa réversibilité a été démontrée chez la souris. Dans le cadre du syndrome de Rett cette thérapie se heurte aux difficultés suivantes:
Il existe deux types de vecteurs utilisés pour les essais de thérapies géniques : les liposomes (vésicules de lipides contenant de l’ADN nu) et les virus. Les premiers présentent une possible toxicité et la pénétration cellulaire est faible. Les virus ont une meilleure pénétration cellulaire mais peuvent contenir qu’une quantité limitée d’ADN (ils contiennent par exemple uniquement le gène d’intérêt si celui-ci n’est pas trop long mais fort heureusement c’est le cas avec MECP2). Ils peuvent néanmoins produire une réaction immunitaire mettant en péril l’action effective de la thérapie génique (cas où la patiente a déjà été en contact avec le vecteur viral).
Jean-Christophe Roux revient sur les travaux majeurs qui ont montré la réversibilité des symptômes chez la souris par introduction du gène MECP2 en thérapie génique. On pourrait donc, en théorie, intervenir à tout moment / tout âge pour guérir une personne atteinte du syndrome de Rett. Ces travaux pré-cliniques chez la souris ont été menés préventif par injection du gène directement dans le cerveau à la naissance chez des souris modèles du syndrome de Rett. Malheureusement ces superbes résultats ont été obtenus de manière préventive avant même que la pathologie ne soit installée. Par la suite, il a été montré qu’une injection intraveineuse chez ces mêmes souris modèles plus âgées (présentant la pathologie), donne des résultats moins convaincants avec une amélioration clinique beaucoup moins importante. Par ailleurs, on observe un problème de reproductibilité entre travaux de différentes équipes.
Dans l’équipe de Jean-Christophe Roux à Marseille :
-utilisation du virus AAV9-Mecp2 comme vecteur de thérapie génique chez la souris modèle
-injection à 30 jours (quand les symptômes apparaissent)
-Imagerie pour détecter l’expression de MECP2 dans des tranches de cerveau qui permet de quantifier le taux d’infection, il est faible avec seulement 10% des cellules qui intègrent le gène.
-chez le mâle, malgré ce faible taux, on note une amélioration notable de la condition des souris (poids, survie et apnées).
–chez la souris femelle : on note également une amélioration mais aussi une toxicité cellulaire, avec notamment des anomalies au niveau du foie.
Comment contrer ces effets indésirables ? Il y a deux axes de recherche :
Méthode alternative testée à Marseille : travail avec la BDNF, une « super-vitamine » neuronale qui favorise la connectivité neuronale. On sait que chez les patientes Rett les neurones sont plus petits et moins bien connectés et on sait également que la BDNF est en quantité réduite chez ces même patientes (quand MECP2 n’est pas présent). Les chercheurs étudient donc la possibilité d’introduire la BDNF à l’aide d’un vecteur viral dans les cellules du cerveau. Un autre avantage du BDNF (par rapport à MECP2) est qu’il est libéré à l’extérieur de la cellule et a donc qu’il a une action également sur les cellules voisines de celles effectivement infectées par le virus vecteur. L’équipe a obtenu de très bon résultats chez la souris mâle (essais chez la femelle à venir avec un contrôle particulier sur des effets indésirables cités ci-dessus : toxicité et duplication). L’équipe étudie également, en collaboration avec des physiciens, l’utilisation d’ultrasons pour ouvrir temporairement la barrière hemato-encéphalique afin de favoriser le passage du vecteur viral et d’augmenter le taux d’infection des cellules cérébrales par le virus.
Autre approche d’intérêt : Réparer le morceau d’ADN muté par la technique des Ciseaux moléculaires (Crispr/Cas9). Cette technique marche uniquement pour certains types de mutation et elle fonctionne très bien en boite de Pétri mais elle est difficile à utiliser actuellement in vivo car on a besoin de 2 vecteurs viraux, un pour les ciseaux moléculaires et un pour réparer le gène sain. Il faudrait donc être sûrs que les 2 vecteurs infectent strictement les mêmes cellules).
Place aux questions : discussion autour du prix du traitement de thérapie génique proposé par une compagnie privée mis en stand-by (très cher) ; effets de traitements sur la microcéphalie chez la souris ? -Eventuellement une légère croissance mais rien de prouvé ; pourquoi les symptômes apparaissent ou évoluent à certaines périodes ? – on pense que c’est lié à l’importance du gène MECP2 dans certaines phases de développement où le cerveau s’organise ; Naît-on malade ?- il semble que de faibles anomalies biologiques liées à des mutations dans MECP2 peuvent être détectées dès la naissance mais elles seraient compensées jusqu’à un certain stade de développement.
Quid de la mention ‘Retard mental sévère’ sur le site de l’AFSR ? Site web CAApable ; discussion autour des logiciels de communication, commande oculaire, PODD sur ordi, Mindexpress…

Accédez à la vidéo diffusée lors de cette conférence
Susan Norwell est une des co-fondatrices de Rett University et possède plus de 40 années d’expérience professionnelle dont plus de 30 consacrées au syndrome de Rett.
En véritable passionnée, elle a pu travailler tout au long de ces années auprès de plus de 300 personnes atteintes du syndrome de Rett. Si Susan est certaine d’une chose, c’est que si l’on est convaincu de leurs compétences, les possibilités sont étendues.
La vidéo rassemble des messages et des conseils illustrés par des vidéos, des instants de travail entre Susan Norwell et des enfants atteintes du syndrome de Rett judicieusement sélectionnés.
Ainsi, elle a partagé, à travers ce film enregistré spécialement pour ces 31èmes Journées Nationales du Syndrome de Rett, ce qu’elle a appris des personnes présentant un syndrome de Rett sous la forme de son « Top 10 » :
Aller plus loin:
http://www.focusedlearningsolutions.com
http://rettuniversity.org/flipbookdemo/
 Accédez à la vidéo diffusée lors de cette conférence
Accédez à la vidéo diffusée lors de cette conférence
Meir Lotan M.Sc.PT Ph.D. est kinésithérapeute et Professeur d’Université à Tel Aviv.
Il a travaillé avec des filles atteintes du syndrome de Rett au cours des 26 dernières années. Il fait partie de l’équipe nationale d’évaluation d’Israël et a reçu en consultation environ 500 filles atteintes du syndrome de Rett à travers le monde. Il travaille comme physiothérapeute avec 15 filles chaque semaine. Il a écrit 30 articles et 4 livres sur le syndrome de Rett et a mené de nombreux programmes de recherche sur ce sujet.
La vidéo nous présente l’évaluation de Sophia, une petite fille de 6 ans, atteinte du syndrome de Rett et marchante (en ‘bonne condition physique’).
Messages / conseils clés : préserver sa motricité et stimuler l’utilisation de ses mains par des exercices à la fois actifs et passifs, en alternance. Augmenter la durée des exercices graduellement.
Autres conseils délivrés :
Aller plus loin:
https://www.ariel.ac.il/wp/physiotherapy/en/
https://scholar.google.com/citations?user=p0mI_RgAAAAJ&hl=en

Le Professeur Laurent Villard est directeur de recherche à l’INSERM de Marseille au sein de l’équipe Neurogénétique Humaine, dont les deux thèmes d’études principaux sont le syndrome de Rett et les encéphalopathies épileptiques précoces. Il a présidé le Conseil Médical et Scientifique de l’AFSR pendant de nombreuses années.
Il a abordé lors de cet atelier, à travers de courtes vidéos très ludiques, les notions de génome, ADN, mutation et gène. Le corps humain est constitué de milliards de cellules issues d’une première cellule qui s’est divisée pour en produire de nouvelles. Dans chacune d’entre elles, il y a un noyau qui renferme toute notre information génétique, répartie sur 46 chromosomes (23 paires) constitués d’ADN. L’ADN est formé de deux brins enroulés l’un autour de l’autre pour former une double hélice. Chacun de ces brins est constitué de quatre nucléotides, A, C, G et T (Adénine, Cytosine, Guanine ou Thymine) liés entre eux formant ainsi une chaîne. L’ordre dans lequel se succèdent les nucléotides le long d’un brin d’ADN constitue la séquence de ce brin. C’est cette séquence qui porte l’information génétique. Celle-ci est structurée en gènes qui ne constituent que 2 % de tout l’ADN. La succession des bases nucléiques sur les gènes de l’ADN détermine la succession des acides aminés qui constituent les protéines issues de ces gènes. La correspondance entre bases nucléiques et acides aminés s’appelle le code génétique. L’ensemble des gènes d’un organisme constitue son génome.
Le génome est en quelque sorte un plan détaillé de notre corps avec des instructions qui permettent son bon fonctionnement. Á la lecture de ce « plan », le corps produit tout ce dont il a besoin pour grandir, se développer, se défendre des agressions extérieures : en un mot, pour vivre !
Depuis que l’on a commencé à séquencer le génome humain, on s’est rendu compte que chaque individu est unique sur le plan génétique (avec 0,1 % de différence entre deux individus soit trois millions de variants génétiques), c’est pourquoi on parle plus souvent maintenant de « variants » plutôt que de « mutations ». Un classement des variants a été défini et détermine cinq types de variants : variant bénin, probablement bénin, de signification inconnue, probablement pathogène, pathogène.
Didier Schwab, maître de conférences à l’Université de Grenoble

Accéder au diaporama présenté lors de cette conférence
L’atelier commande oculaire et logiciel libre de droit1 a été conduit par Didier Schwab. Il se présente à double titre comme le papa de Tess, 7 ans, porteuse du Syndrome de Rett et en tant que chercheur en informatique. Actuellement maître de conférences, attaché à l’université de Grenoble, ses travaux se concentrent autour de 4 axes regroupant l’informatique, la cognition, les interactions Homme-Machine et la linguistique. Autrement dit comment communiquer grâce à la machine en contexte multilingue ou avec des personnes à besoins spécifiques notamment avec la Communication Augmentée Alternative – CAA.
Il existe différents types d’outils CAA et de techniques : objets tangibles, gestes, pictogrammes, logiciels de synthèse vocale, oculomètres (ou eye-tracker).
Choisir un outil de CAA pour une personne polyhandicapée ; c’est se poser des questions sur la pertinence du contenu de l’outil sans négliger son ergonomie à savoir la manière dont l’outil pourra être manipulé. L’utilisation du regard afin d’interagir avec l’environnement est souvent considéré pour une personne polyhandicapée comme l’un des moyens de communication les plus naturels et faciles à mettre en œuvre. Néanmoins l’enfant devra apprendre le pouvoir de ses yeux, comprendre les conséquences des actions déclenchées par son regard et entraîner ses muscles extra-oculaires. C’est par le biais de jeux adaptés que l’enfant peut faire cet apprentissage destiné à permettre l’utilisation de logiciels de communication utilisant des pictogrammes.
Didier Schwab et son équipe ont développé des jeux libres et gratuits regroupés sous le site http://Gazeplay.net. La soixantaine de jeux actuellement proposés sont compatibles avec n’importe quel oculomètres, ils sont en source ouverte (open source) afin de développer plus facilement une communauté (parents, aidants, institutions spécialisées) et assurer une pérennité. L’Utilisation de Java facilite son développement sur de nombreuses plateformes. Il est gratuit pour le rendre accessible au plus grand nombre. Les Jeux ont été conçus grâce aux retours d’aidants, de professionnels, d’étudiants et initialement de 3 petites filles Rett
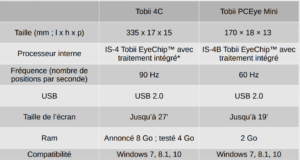
Le regard de la personne est capté par un oculomètre (eye-tracker), qui va en quelque sorte remplacer la souris. Il s’agit d’une sorte de barrette longiligne qui se fixe au bas d’un écran d’ordinateur. Il s’agit d’une technologie déjà ancienne utilisée depuis plus de 30 ans dans le domaine du handicap. Des eye-trackers destinés au monde du jeux vidéo (Tobii 4C par exemple) et aux caractéristiques similaires à ceux du monde du handicap (Tobii PCEyeGo par exemple) existent.
Le tableau ci-dessous en présente une comparaison :
![]()
Selon l’American Speech Language Hearing Association, la CAA répond aux besoins des individus avec des troubles de communication importants et complexes qui sont caractérisés par des déficiences dans le langage. La communication est augmentée pour compléter un langage préexistant, elle est alternative en remplacement d’un langage non existant ou dysfonctionnel.
Certains logiciels CAA sont directement compatibles avec les eye-trackers comme GazeSpeaker (gratuit) ou GazePlay. D’autres nécessitent de passer par la souris : Gaze Mouse (libre et gratuit) ou chez Tobii GazePoint , gratuit et largement suffisant pour les personnes porteuses du syndrome de Rett. Il permet de contrôler un dispositif windows (sur ordinateur, ordinateur tactile chevalet, ordinateur fixe avec chariot mobile ou tablette) avec 4 Go de RAM.
Le matériel et les logiciels ne sont plus un problème financier c’est avant un problème de formation et de sensibilisation des parents, des aidants, des professionnels…
Contact : gazeplay.net@gmail.com
Pour ses 31èmes Journées Nationales du Syndrome de Rett, l’AFSR remercie chaleureusement l’ensemble des participants, ainsi que :
Dr Jean-Christophe Roux, M. Luc Rivoira, Mme Sabine Blanchard, Mme Mélanie Toulouse, Mme Myriam Fieschi, Mme Sabrina Tramaille-Bryon, Pr Raphaël Vialle, Pr Brice Illharreborde, Dr Bénédicte Pigneur, Pr Laurent Villard, Pr Mathieu Milh, Mme Jeanne Mendelssohn, M. Didier Schwab pour leurs interventions durant ce week-end.
Susan Norwell et Meir Lotan pour avoir enregistré ces vidéoconférences à l’occasion de notre événement.
Les fondations Apicil, Groupama pour la Santé, EDF, la société Inédit Immobilier, ainsi que la direction d’Eurosport et l’ensemble de son personnel pour leur soutien financier.
Toute l’équipe adresse un remerciement spécial aux bénévoles qui ont répondu présents pour nous apporter aide et soutien lors de ce week-end ainsi qu’à l’élaboration des comptes-rendus.
Á tous, un immense merci !

![]()


Association Française du Syndrome de Rett
264 rue du Champ Monette
60600 AGNETZ
09.72.41.09.89
contact@afsr.fr
Contrairement au Séjour Familles, cet événement ne bénéficie pas de dispositions particulières pour accueillir les personnes atteintes du syndrome de Rett.
Ligne 13 : Station BROCHANT
Ligne 13 : Station PORTE DE CLICHY
Ligne 31, 54, 66, 74 :Station BROCHANT
Ligne PC3 : Station PORTE DE CLICHY
Ligne C : Station PORTE DE CLICHY
PARIS ST LAZARE (3.00 km / 1.86 mi)
PORTE DE CLICHY (0.10 km / 0.06 mi)
Vous pouvez bénéficier de 20% de réduction sur votre billet SNCF sur simple demande à contact@afsr.fr.
CHARLES DE GAULLE AEROPORT (20.00 km / 12.43 mi)
PARIS ORLY (25.01 km / 15.54 mi)
LE BOURGET AEROPORT (17.00 km / 10.56 mi)
Sortie périphérique Porte de Clichy, entrer dans Paris, prendre l’avenue de Clichy, 1ère rue sur la droite au bout de 200m, Rue Bernard Buffet.
Dans le cas où votre venue en transport en commun est impossible, vous pourrez bénéficier d’une place de parking au sein de l’hôtel prise en charge par l’AFSR.
Novotel Paris Charenton Le Pont
3-5 place des Marseillais
94227 Charenton Le Pont



Informations sur le parking :
Source : https://all.accor.com/hotel/1549/index.fr.shtml
- Sur place
- Parking intérieur
- Privé
- Tarif du parking : 20.00€ Par jour
Les 29èmes Journées Nationales du Syndrome de Rett de l’AFSR se sont déroulées les 7 et 8 octobre 2017 à l’hôtel Ibis Paris 17 Clichy-Batignolles et ont accueilli près de 230 personnes.
Cet événement dédié au syndrome de Rett, et plus largement au polyhandicap, était ouvert aux familles, mais aussi pour la première fois, aux proches ou aidants d’une personne atteinte du syndrome de Rett, aux professionnels de santé, scientifiques, personnels de centres spécialisés, et étudiants, à tarif préférentiel.
Un vaste programme de conférences a été proposé aux participants durant deux jours. L’Association Française du Syndrome de Rett remercie chaleureusement l’ensemble des professionnels qui ont répondu présent pour animer ces conférences.
Pour consulter le résumé des 29èmes JNSR : cliquez ici (pages 2 à 4)
Le programme était le suivant :
 |
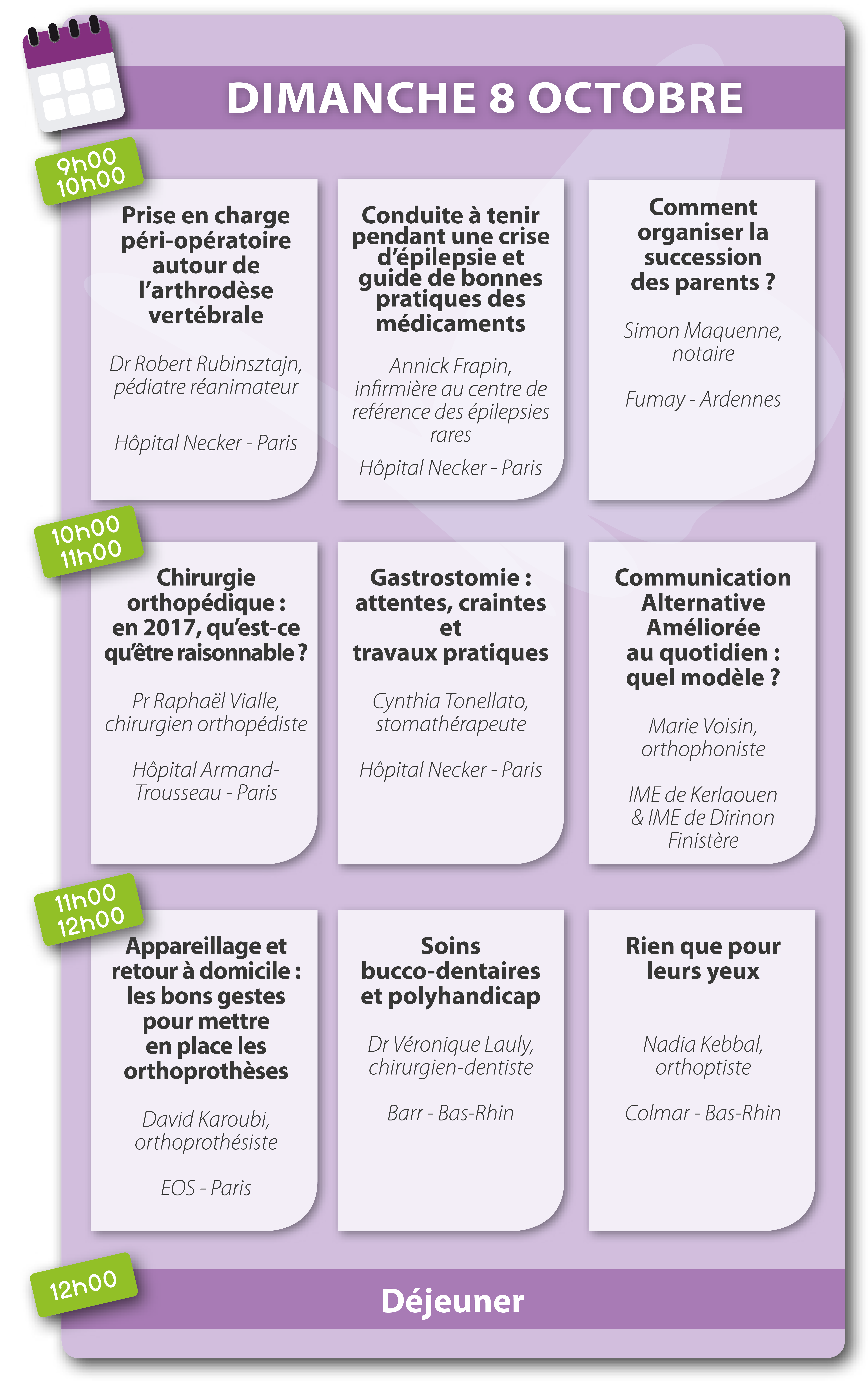 |
Les 29èmes JNSR ont été organisées par l’AFSR et avec le soutien de :
Les 30èmes Journées Nationales du Syndrome de Rett de l’AFSR se sont déroulées les 6 et 7 octobre 2018 à l’hôtel Ibis Paris 17 Clichy-Batignolles et ont accueilli plus de 230 personnes.
Cet événement dédié au syndrome de Rett, et plus largement au polyhandicap, était ouvert aux familles, mais aussi pour la première fois, aux proches ou aidants d’une personne atteinte du syndrome de Rett, aux professionnels de santé, scientifiques, personnels de centres spécialisés, et étudiants, à tarif préférentiel.
Un vaste programme de conférences a été proposé aux participants durant deux jours. L’Association Française du Syndrome de Rett remercie chaleureusement l’ensemble des professionnels qui ont répondu présent pour animer ces conférences.
Pour consulter le résumé des 30èmes JNSR : cliquez ici
Le programme était le suivant : cliquez ici
Les 30èmes Journées Nationales du Syndrome de Rett sont organisées par l’Association Française du Syndrome de Rett et avec le soutien de :
![]()

![]()
![]()






























